Dilated cardiomyopathy (DCM) is the most common type of cardiomyopathy and one of the leading causes of heart failure (HF). Several mutations identified in phospholamban (PLN) have been linked to familial DCM and HF, yet the underlying molecular mechanism remains controversial. Under physiological conditions, PLN interacts with sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) and regulates calcium uptake, which is modulated by the protein kinase A (PKA)-dependent phosphorylation of PLN during the fight-or-flight response. To reveal the pathological mechanism of cardiomyopathy caused by genetic mutations in PLN, the research group of Zhiguang Yuchi of Tianjin University published a research article entitled “Structures of PKA-phospholamban complexes reveal a mechanism of familial dilated cardiomyopathy” in eLife. They solved the protein crystal structures of the catalytic domain of mouse PKA in complex with wild-type and DCM-mutant PLNs. The structures, combined with the results from other biophysical and biochemical assays, reveal a common disease mechanism: the mutations in PLN reduce its phosphorylation level by changing its conformation and weakening its interactions with PKA. In addition, they demonstrate that another more ubiquitous SERCA-regulatory peptide, called another-regulin (ALN), shares a similar mechanism mediated by PKA in regulating SERCA activity.

25-35% of DCM cases have familial origin, caused by inherited mutations in genes encoding proteins involved in muscle contraction and calcium handling, including PLN [1-4]. PLN, an important regulator of SERCA, is a 6.2 kDa single-pass integral membrane protein that can reversibly inhibit SERCA activity by physically interacting with the calcium pump and thus regulate the contraction of cardiac muscle. Inhibition of SERCA by PLN responds to changes in the free calcium concentration and redox environment of the cytosol. The potency of SERCA inhibition also depends on certain structural properties of PLN, such as its oligomerization state and phosphorylation levels [5-6]. To date DCM mutations associated with PLN include R9C, R9H, R9L, ΔR14, R14I, and I18T [7-10]. Despite of extensive study, their disease mechanisms are far from clear.
The authors determined the protein crystal structure of PKA in complex with wild-type and two mutant PLNs. Compared with the previously solved structure of the wild-type complex with a ratio of PKA:PLN=2:1, the current structure of the wild-type complex with a ratio of PKA:PLN=1:1 is closer to its real solution form, revealing how PKA catalyzes PLN substrates under physiological conditions. The crystal structure of the PKA+PLN A11E complex solved by the team further distinguished the two wild-type structural models and confirmed the correctness of the newly solved wild-type structure. R9C is the most severe phenotype and the most widely studied DCM-related mutation, but the molecular mechanism of how it causes cardiomyopathy remains controversial due to lack of structural information. The team further solved the crystal structure of the complex of PKA and PLN R9C, showing that R9C abolished an electrostatic interaction between the positively charged side chain of arginine and the negatively charged α-helical dipole at the PKA binding pocket , and another hydrogen bond with PKA, which causes PLN to change conformation and move away from PKA, ultimately weakening the interaction between the two (Figure 1).
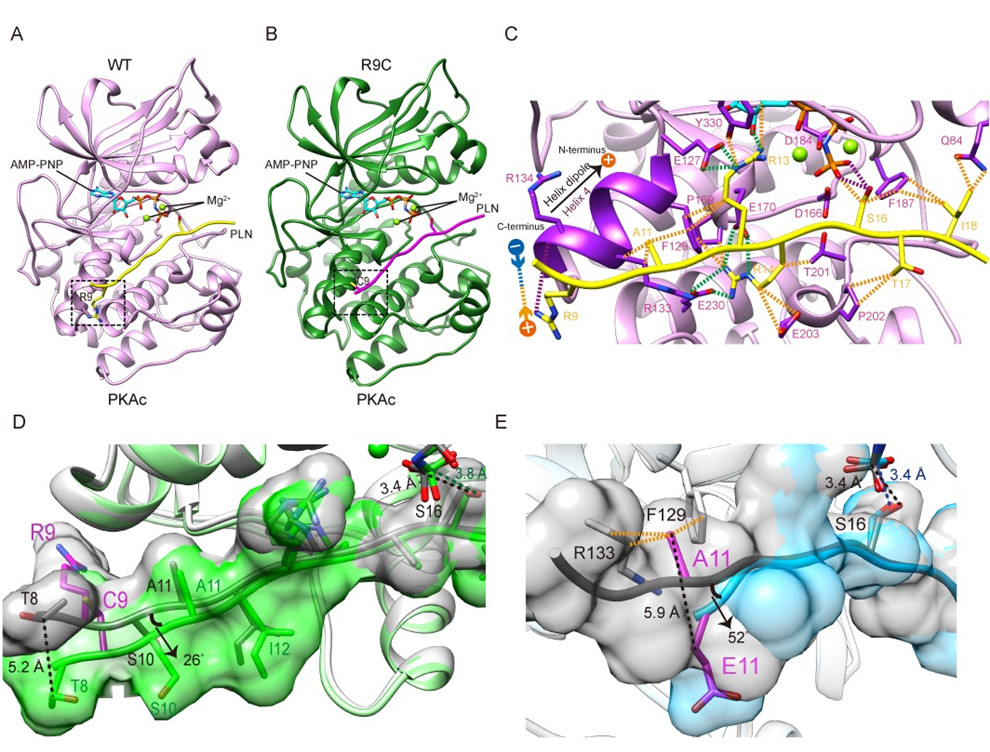
Fig. 1. Crystal structures of PKAc in complex with wild type and mutant PLNs.
To verify whether the DCM mutations affect the binding between PKA and PLN, the team characterized the interaction between the two and measured the enzyme kinetic constants through surface plasmon resonance technology, thermostability experiments, ADP-Glo kinase experiments, and NMR. These mutations do reduce the catalytic efficiency of the kinase by altering its conformation and attenuating the interaction between PLN and PKA, thereby achieving attenuated phosphorylation levels of PLN. In addition to disease mutations, the authors found that PLN phosphorylation mediated by calmodulin-dependent protein kinase II (CaMKII) also attenuates the interaction between PLN and PKA, and reduces the phosphorylation level at the PKA site on PLN, demonstrating that there is a crosstalk between two signal pathways (Figure 2).
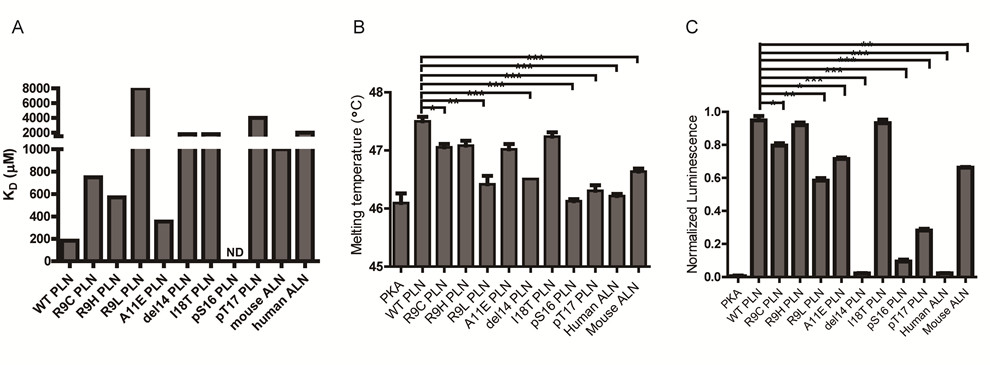
Fig. 2. (A) Surface plasmon resonance; (B)Thermal shift assay; (C) ADP-Glo kinase assay.
To find out whether the mechanism of phosphorylation regulation of SERCA regulatory peptides is conserved, the authors conducted similar studies on a newly identified SERCA regulatory peptide, ALN. ALN is a single-pass transmembrane protein expressed on the endoplasmic reticulum membrane and is widely expressed in many tissues and organs including the cardiomyocyte. The transmembrane domain of ALN responsible for the interaction with SERCA is relatively conserved in sequence with PLN, while the sequence of the potential phosphorylation regulatory site on the cytoplasmic side is not conserved. The experimental results showed that ALN could be phosphorylated by PKA, however, the binding affinity of ALN to PKA and its phosphorylation level were decreased compared with PLN.
This study provides an important theoretical basis for understanding the pathological mechanism of DCM, and also provides important clues for understanding the physiological function of PLN in the calcium signaling pathway of muscle cells and the general catalytic mechanism of PKA.
Prof. Zhiguang Yuchi and Prof. Yan Zhang from the school of pharmaceutical science and technology (SPST) of Tianjin University are the co-corresponding authors of the paper. Juan Qin, a PhD student from SPST, is the first author. Prof. Jingfeng Zhang from Wuhan Institute of Physics and Mathematics, Chinese Academy of Sciences, Prof. Zhi Lin, Prof Kenneth J Woycechowsky and Ph.D. student Lin Lianyun from Tianjin University, Prof. Filip Van Petegem and Dr. Omid Haji-Ghassemi from University of British Columbia made important contributions to this project. The project is supported by the National Natural Science Foundation of China for Excellent Young Scholars and the Natural Science Foundation of Tianjin. The project was also supported by the X-ray Diffraction Facility of Shanghai Synchrotron Radiation Facility (SSFR).
The research group of Prof. Yuchi of Tianjin University (www.yuchilab.com) focuses on the structural and functional study of important drug targets and the screening and development of drugs. We are recruiting doctoral students and postdoctoral fellows, and sincerely invite talents from all over the world to join! (Email: yuchi@tju.edu.cn)
Reference:
1. Alves, M.L., R.D. Gaffin, and B.M. Wolska, Rescue of familial cardiomyopathies by modifications at the level of sarcomere and Ca2+ fluxes. J Mol Cell Cardiol, 2010. 48(5): p. 834-42.
2. MacLennan, D.H., Ca2+ signalling and muscle disease. Eur J Biochem, 2000. 267(17): p. 5291-7.
3. MacLennan, D.H. and E.G. Kranias, Phospholamban: a crucial regulator of cardiac contractility. Nat Rev Mol Cell Biol, 2003. 4(7): p. 566-77.
4. Kranias, E.G. and D.M. Bers, Calcium and cardiomyopathies. Subcell Biochem, 2007. 45: p. 523-37.
5. Ha, K.N., et al., Lethal Arg9Cys phospholamban mutation hinders Ca2+-ATPase regulation and phosphorylation by protein kinase A. Proc Natl Acad Sci U S A, 2011. 108(7): p. 2735-40.
6. Yu, X. and G.A. Lorigan, Secondary structure, backbone dynamics, and structural topology of phospholamban and its phosphorylated and Arg9Cys-mutated forms in phospholipid bilayers utilizing 13C and 15N solid-state NMR spectroscopy. J Phys Chem B, 2014. 118(8): p. 2124-33.
7. Schmitt, J.P., et al., Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science, 2003. 299(5611): p. 1410-3.
8. Medeiros, A., et al., Mutations in the human phospholamban gene in patients with heart failure. Am Heart J, 2011. 162(6): p. 1088-1095 e1.
9. Burns, C., et al., Multiple Gene Variants in Hypertrophic Cardiomyopathy in the Era of Next-Generation Sequencing. Circ Cardiovasc Genet, 2017. 10(4).
10. Schmitt, J.P., et al., Alterations of phospholamban function can exhibit cardiotoxic effects independent of excessive sarcoplasmic reticulum Ca2+-ATPase inhibition. Circulation, 2009. 119(3): p. 436-44.
Full-text link:https://elifesciences.org/articles/75346
